Experience at Niche has taught us that clinical study protocols are born in the white-hot fire of new asset development programmes, where the focus is on driving forward. Under these often‑frenetic conditions there is an increased risk of oversight and/or error, particularly in the process of clinical study design and subsequent protocol development. Being a risk‑sensitive industry, study design and protocol development is an iterative process, with plans often being modified several times before we even commit pen to paper [2]. This belt‑and braces approach is augmented during the early stages by the use of concept protocols, we have discussed their judicious use previously in an issue of our Insider’s Insights.
The final protocol is a blueprint for investigators and clinical delivery teams to provide the study, and its development is a skill comparable with an experienced architect preparing the plans for a landmark development. It requires contributions from a plethora of speciality departments and outlines the future budget spend, potentially running into the tens of millions. Any necessary modifications are managed with dexterity through clearly documented protocol amendments. The implications of protocol changes to study delivery and the overall study budget may be considered modest until we enter the clinical domain and start to operationalize the study.
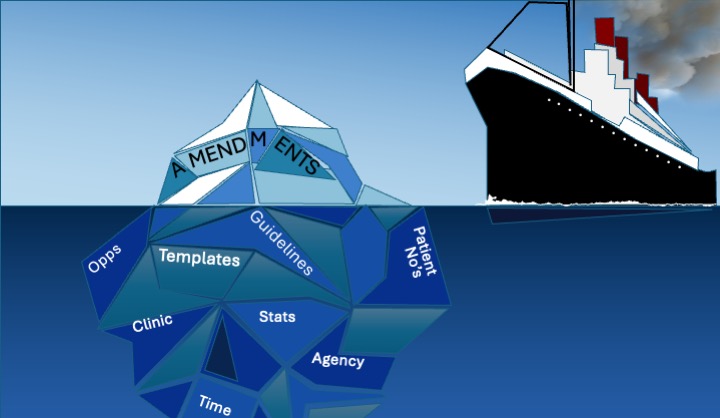
Protocol amendments are generally accepted as to be a fact of life in clinical trials. We deal with them. But does our ‘reactive’ approach leave u simply dealing with the tip of the iceberg. Are we steaming ahead without giving due consideration to something much larger lurking below the surface? Even protocols for studies employing the simplest designs will see 2–3 amendments during their lifetime [1][3]. Modifications to the protocol can have significant implications. A widely cited report from 2011 suggests that a single protocol amendment can take up to 2 months to deliver and can cost up to $0.5 million – and this Estimate does not account for Sponsor time, spent on implementing the amendment, undertaking necessary translations or regulatory agency resubmissions [1].
Clearly, the financial burden of amendments are greater in later stages of drug development but will always incur some form of change to investigator site fees as well as contract change orders with contract research organisations (CROs) and other third part providers. In fact, the evidence shows that even early protocol modification are not as trivial as generally viewed by the industry. The 2011 analysis suggested (from a very crude analysis) that ‘avoidable’ protocol amendments cost sponsors (in total) as much as $2 billion a year irrespective of possible aggregate time‑savings if they could be minimised [1][4]. So, what does experience teach us of how this can be avoided?
Our own experience over the last 20 years at Niche seems to concur with the often held belief that most frequently occurring reasons for amendments are:
These causes reflect a wide range of choices that clinical study teams must consider to manage overall performance. They also reflect judgement calls, where a risk‑based approach has been adopted by clinical research management in order to reach ever tighter delivery deadlines and budget restrictions. The challenge of protocol complexity and unforeseen operational issues has been widely documented in the literature [5].
We have found that attempts to develop better study designs that incorporate elements of flexibility to address ever‑increasing protocol complexity, often reflected in adaptive study design protocols, serve to reduce the need for amendments [6]. Overall, techniques that appear to yield substantial cycle time and budget savings are:
Clear consideration of recruitment targets, feasibility assessments and retrieval strategies that anticipate problems and can minimise delay and risk of failure to hit targets. Review of potential study population is particularly important as nearly 20% of all studies in patients require changes to the subject eligibility criteria [7]. Include clinical study manuals to keep study protocols as simple as possible avoiding unnecessary duplication of information (particularly in terms of timing of procedures). Employ adaptive study designs to provide flexibility in study delivery without foreseeable need for protocol amendment.
Despite the continuing impact that unnecessary protocol amendments have on delivery timelines and study budgets the current industry model for protocol development appears to contradict what we know of best practice. The market dominance of global CROs has seen the ‘writing’ function of clinical study conduct diverted to large medical writing teams.
Although this has seen overall improvements in the quality and consistency of regulatory documents, the delivery model has inherent limitations.
Writing teams may not always be based in the country where the study is to be conducted and writers may not therefore be aware of local regulations. In addition, use of writing team resources within large CROs is usually rationalised to ensure their efficient (and profitable) exploitation. Finite in nature, these resource allocations are often planned‑out using known delivery schedules – for example, when tables, figures and listings become available for writing a clinical study report, when the toxicity data will be reported when preparing an Investigator’s Brochure or when finalised protocol is available for the IMPD and CTA submission. This contrasts with the more complex nature of clinical protocol delivery.
By their very nature, clinical protocols and amendments do not fit efficiently in these planning models. Sponsors want a first draft of a protocol as soon as possible, where possible within 5–10 days of signing a new contract or Task Order, and amendments within a couple of days. This can leave the CRO scrabbling around for resource, solutions often see the main protocol author lacking expertise, operational experience and/or therapy area understanding, leaving your draft protocol open to the errors detailed above.
These challenges have been known for some time but there is little evidence that the industry has improved its performance since 2011. A Tufts Center for the Study of Drug Development analysis performed in 2016 indicated that nearly 60% of protocols may experience at least one substantial amendment during their lifetime, having a significant impact on study duration [1][3].
It is true that clinical professionals see amendments to protocols as a necessary evil but are the often simply a consequence of inappropriate review and poor planning? Of the amendments made, it seems that nearly 40% occur before the first study volunteer receives their first dose, particularly in Phase I. This seems an unfortunate use of time and resource. Hopefully the information summarise here will help us all to focus on how we can do better.
References

Get our latest news and publications
Sign up to our news letterResources
Social


Contact us
Address
Niche Science & Technology
Unit 26 Falstaff House
Bardolph Road
Richmond TW9 2LH
United Kingdom
