For sometime now (decodes!) I have watched the pharmaceutical industry adopt technological innovation for the process of drug discovery, clinical data management, biostatistics and pharmacovigilance. Yet one critical component of the drug development pathway remains remarkably dependent on manual effort: the preparation of regulatory documents.
Among these documents, the Clinical Study Report (CSR) occupies a central role. Required for regulatory submissions worldwide, the CSR provides a comprehensive account of the design, conduct, analysis and interpretation of a clinical trial. It represents the definitive record of a study and forms a critical component of the evidence package supporting decisions regarding drug approval and clinical use [1].
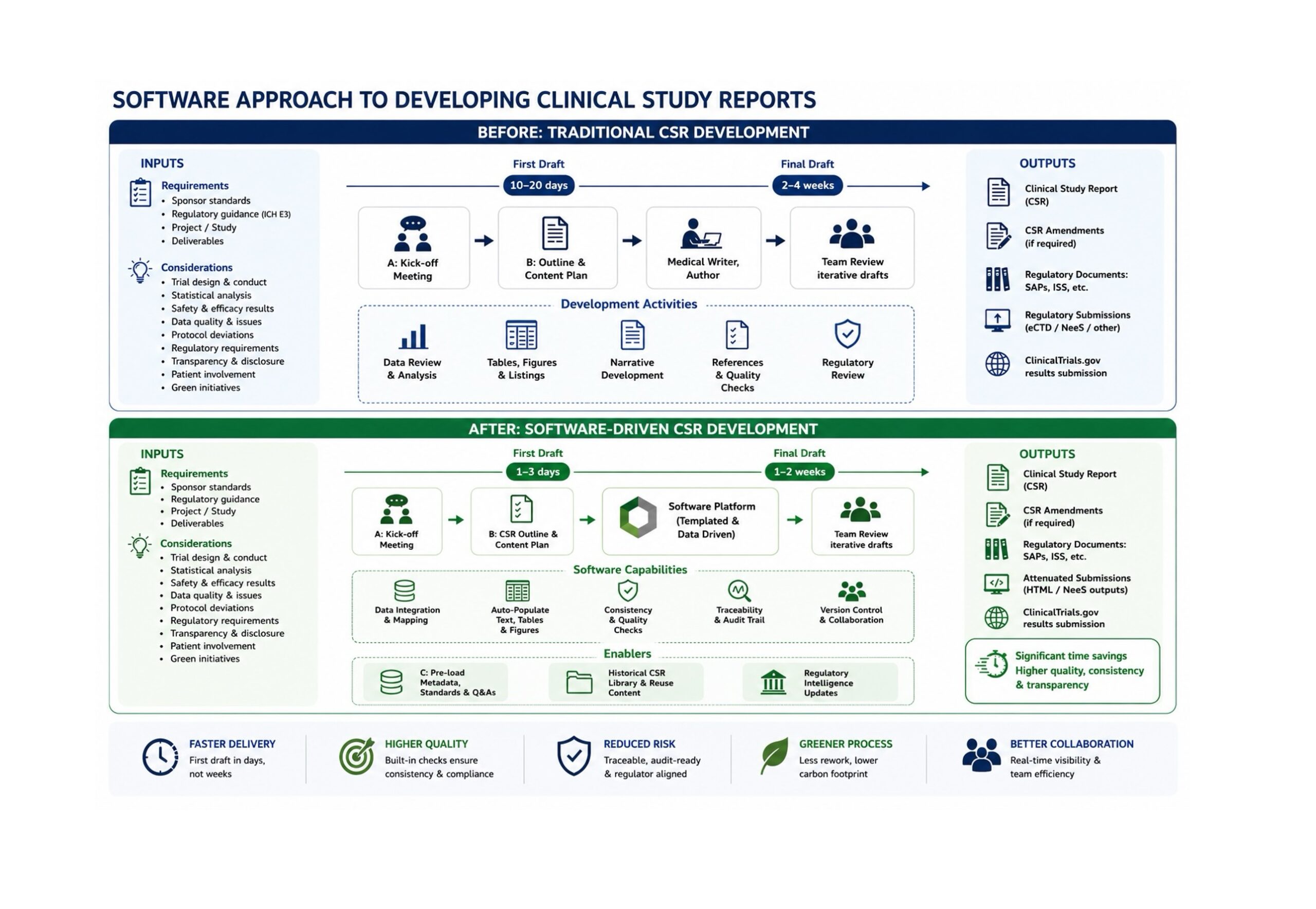
Despite advances in electronic data capture, clinical databases and statistical programming, the production of CSRs remains largely a labour-intensive process involving multiple stakeholders, extensive document review cycles and significant manual assembly of information. When done successfully, this process is usually managed by professional medical writers who will; coordinate the various data sources from different disciplines. Although a highly skilled occupation, report writing represents a great opportunity for saving. For this reason, Niche Science & Technology (NST) has submitted a grant proposal to the UK Technology Strategy Board’s Biomedical Catalyst programme to investigate whether this process could be fundamentally transformed through software-assisted automation.
Our proposal is founded upon a simple but potentially disruptive observation: if clinical trial data are already generated electronically, and if regulatory reporting follows a highly standardised structure, why should creation of the final report continue to depend wholly on months of manual compilation?
The development of a modern pharmaceutical product generates vast quantities of documents and data. Protocols, statistical analysis plans, patient data listings, efficacy analyses, safety summaries and quality assurance records must ultimately be consolidated into regulatory documents that meet stringent international standards.
The structure of the CSR has been largely standardised since publication of the International Conference on Harmonisation (ICH) E3 guideline in 1995 [1]. The guideline established a common framework that allows regulators to efficiently review clinical evidence across studies and therapeutic areas. While this standardisation has improved consistency, it has also highlighted the repetitive nature of many reporting activities.
Medical writers frequently spend considerable time transferring data from statistical outputs into predefined report sections, ensuring consistency across tables and narratives, and checking that information is accurately represented throughout the document. Although scientific judgement remains essential, many components of the process involve structured information management rather than scientific interpretation.
The increasing complexity of clinical development programmes and individual trials has amplified this challenge. As we entered the 2010s, concerns regarding escalating drug development costs had become a major focus of industry discussion [2,3]. Estimates suggested that bringing a new medicine to market could require investments exceeding a billion dollars when accounting for failures and opportunity costs [2]. Any activity capable of reducing development timelines have thus attracted considerable attention. Whereby, less time equals less money
Regulatory documentation represents one such opportunity.
Our proposal has emerged from practical experience. By the start of 2013, the Niche team had been involved in delivery of over 500 Clinical Study Reports, during the process accumulating extensive expertise in medical writing, clinical project management and regulatory consultancy. Through this experience, we identified numerous aspects of CSR preparation that appeared suitable for automation.
Our proposal argues that much of the information required within a CSR already exists in electronic form. Clinical databases contain patient-level data. Statistical software generates tables, listings and figures. Study management systems hold protocol information and operational records. Yet these data sources are rarely integrated into a unified reporting process. Instead, information is often manually extracted, reformatted and inserted into document templates. Every transfer introduces opportunities for inconsistency, transcription errors and delays if not managed professionally.
We have proposed developing a software platform capable of extracting information automatically from existing data sources and populating validated CSR templates. Rather than replacing scientific expertise, the system would eliminate repetitive manual activities and augment the skills of operators. allowing writers to focus on interpretation, team consensus (on findings), quality control and scientific communication.
Our ambition is substantial. The proposal envisions reducing report preparation timelines from months to days while simultaneously improving consistency and reducing resource requirements.
Not all regulatory documents are equally suitable for automation.
The CSR presents a particularly attractive target because of its highly structured nature. The ICH E3 guideline defines the major sections of the report, including study objectives, methodology, patient disposition, efficacy analyses, safety findings and conclusions [1]. Although scientific interpretation varies between studies, much of the document follows predictable patterns. This standardisation creates opportunities for software-driven document generation.
The proposal recognises that many CSR components can be ‘assembled’ automatically from predefined rules and data mappings. Demographic summaries, disposition tables, efficacy analyses and safety summaries already originate from statistical outputs. Study conduct information frequently resides in project databases and protocols/protocol amendments. Administrative details are typically standardised across studies.
If these elements could be extracted and assembled automatically, substantial efficiencies would be achieved.
Importantly, we have viewed the CSR as only the beginning. The proposal suggested that successful development of automated CSR generation could eventually be extended to investigator brochures, clinical overviews and other regulatory submissions.
The concept of computer-assisted document generation was not entirely new. Automated report generation had already been explored in areas such as weather forecasting, financial reporting and medical diagnostics [4,5]. Similarly, electronic document management systems had become increasingly common within pharmaceutical organisations.
However, the application of these principles to regulatory medical writing remained relatively limited. We identified only a small number of commercial offerings addressing this space and argued that existing products required substantial manual intervention. The company’s proposal therefore focused not merely on document assembly but on creating a more comprehensive workflow that combined data extraction, document generation, review management and secure delivery.
Our aim is for the platform to operate either as a client-hosted system or through a secure web portal, enabling pharmaceutical companies worldwide to access automated reporting services while maintaining confidentiality and regulatory compliance.
Our grant application demonstrates a realistic appreciation of the technical barriers that would need to be overcome.
One challenge involved extracting data from diverse software environments. Clinical development organisations employ various statistical and database platforms, often using customised workflows across external suppliers. Reliable migration of information between these systems is far from trivial.
Another challenge involves handling unstructured information. While numerical data can often be mapped directly into predefined templates, narrative content frequently requires interpretation and contextualisation. Developing methods for extracting and integrating such information represented an important component of the feasibility study. We hope that our experience with machine learning gained through our working with the Brunel University spin-out, Cardionetics, will bring something truly unique to our solution.
Data security also features prominently. Clinical trial information is among the pharmaceutical industry’s most sensitive assets. Any web-based reporting platform would require robust safeguards to ensure confidentiality, integrity and regulatory compliance. There are also growing concerns over the security of patient data.
Finally, market acceptance can not be assumed. Pharmaceutical organisations tend to adopt new technologies cautiously, particularly when they affect documents submitted to regulatory authorities. Demonstrating reliability, accuracy and cost-effectiveness are therefore be essential.
If successful, our proposed platform could deliver benefits extending beyond individual documents. Faster report completion would accelerate availability of study results and potentially reduce delays between development milestones. Improved consistency could reduce quality control burdens and minimise costly document revisions. Reduced resource requirements might allow highly trained scientists and medical writers to focus on higher-value activities requiring expert judgement. Such efficiencies align with broader efforts to improve productivity within pharmaceutical research and development [3].
The proposal also reflects concerns regarding international competitiveness. Right now, many development activities are shifting toward regions offering lower operating costs. We argue that technological innovation offered a means for European organisations to address geopolitical challenges and remain competitive by delivering greater efficiency without compromising quality.
The concept of transforming structured data into regulatory narratives, automating repetitive writing tasks and integrating disparate information sources has become increasingly relevant as digital technologies continue to evolve.
What distinguishes our proposal is its recognition that regulatory writing is not solely a writing problem. It is fundamentally an information management challenge. By addressing the movement of information rather than simply the creation of text, we are preparing a new way of thinking about regulatory documentation.
Whether software could truly generate a complete Clinical Study Report remains to be proven. However, we envisage that the greatest benefit from our solution will be greatest when operated by well-seasoned operators like the Niche medical writing team. Yet the question itself reflected a growing recognition that the future of drug development would depend not only on scientific innovation but also on innovation in how scientific knowledge is organised, communicated and delivered.
In that respect, our proposal represents more than a software project. It was an early exploration of how technology might reshape one of the pharmaceutical industry’s most established and labour-intensive processes.
References

Get our latest news and publications
Sign up to our news letterResources
Social


Contact us
Address
Niche Science & Technology
Unit 26 Falstaff House
Bardolph Road
Richmond TW9 2LH
United Kingdom
